
通讯作者:Peichen Zhong, Gerbrand Ceder通讯单位:加州大学伯克利分校,劳伦斯伯克利国家实验室人工智能(AI)作为一种发现和优化新型电池材料的潜在工具受到了广泛关注。通过利用海量的实验和计算数据,AI辅助技术可以加速电池材料的设计和合成。尽管如此,当前大多数预测电池性能的机器学习模型主要集中在预测特定化学体系或有限化学空间内的性能,例如对于商用化电池的寿命预测;对于多组分正极材料的表征学习,具备探索性的机器学习模型发展仍显不足。这主要是由于同时优化多个电化学测试参数较为困难(例如倍率、循环性能以及不同测试电压窗口),以及在不同组分空间内高保真数据的稀缺性进一步阻碍了AI在电池领域的充分应用。近几个月以来,AI模型正在加速改变计算材料的研究范式。特别是通过对过去近十年的DFT计算数据进行学习,通用机器学习原子间势能函数在材料模拟领域取得了超群的效果,例如基于Materials Project数据库搭建的M3GNet/CHGNet,以及Google DeepMind搭建的GNoME正被广泛应用于无机材料的发现与设计。在此基础之上,我们很自然地会设想建立一条通往实验数据的通用机器学习之路。为此,我们选择了无序岩盐(DRX)材料作为研究背景。DRX材料由于其构造无需保持层状结构,因此可以使用含高丰度元素的前驱体进行合成,例如Mn和Ti。由于其结构的无序性,DRX正极具有近乎无限的成分设计空间,以及更加复杂的结构-性能关系。如何构造出一个通过给定化学组分和电化学测试条件,预测电化学性能的AI模型,是一个充满挑战且有趣的问题。针对这些挑战,我们开发了DRXNet——一种用于电池正极材料发现和优化的探索性机器学习模型。DRXNet将正极材料化学组分、电化学测试电流密度、工作电压窗口以及循环次数作为输入,来预测若干条放电曲线。通过对过去近五年的实验数据进行挖掘,我们构造了一个丰富的训练数据集,包含14种不同过渡金属元素、19,000多条DRX正极材料实验放电电压曲线。通过对以上数据集进行学习,我们表明该模型能够准确捕捉不同测试条件下的正极电化学性能。作为在多样化化学成分上训练的通用模型,DRXNet为快速识别性能得到优化的新型储能材料提供了一种具有启发性的数据驱动解决方案。 DRXNet旨在构造给定条件下的电压和容量之间的关系映射,例如(1)正极材料化学组分和(2)电化学测试条件。这一思路可以概念化为识别一个函数F该函数将电压状态Vi映射为输出容量状态Qi,同时函数F由电化学参数O有条件地定义,其中O包含了电极化学组分、测试电流密度(倍率)和循环次数。如图所示,我们设计的DRXNet由两部分神经网络组成:(1) 电化学条件网络O:根据正极材料的化学组分和电化学测试信息生成特征向量(2) 状态预测网络F:在给定电化学条件编码O的情况下,将容量近似为电压状态的函数, 我们使用Goodall et al.提出的图神经网络模型Roost对正极材料化学组分进行成分编码。Roost将元素作为图节点,并根据每个元素的分数浓度通过加权消息传递更新元素之间的相关性。图节点初始化使用mat2vec中的元素嵌入向量,以通过mat2vec中所挖掘和包含的文本信息来尽可能多地捕获先验化学信息。
我们使用Goodall et al.提出的图神经网络模型Roost对正极材料化学组分进行成分编码。Roost将元素作为图节点,并根据每个元素的分数浓度通过加权消息传递更新元素之间的相关性。图节点初始化使用mat2vec中的元素嵌入向量,以通过mat2vec中所挖掘和包含的文本信息来尽可能多地捕获先验化学信息。
由于倍率和循环性能本质上受到化学组分的影响,我们通过分层网络结构以及门注意力机制对MLP进行电化学条件编码得到包含倍率信息特征向量:其中 和
和 表示具有不同激活函数的MLP,|| 表示连接操作。
表示具有不同激活函数的MLP,|| 表示连接操作。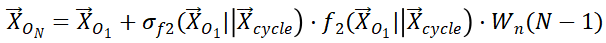
该特征向量与循环次数呈线性相关,通过可训练的权重Wn进行学习。因此,特征向量 用于表示第一个循环,且
用于表示第一个循环,且 可用于表示第N个循环。最后,我们使用MLP构建状态预测网络F,将电压状态Vi和工作窗口 [
可用于表示第N个循环。最后,我们使用MLP构建状态预测网络F,将电压状态Vi和工作窗口 [ ,
, ] 作为输入,通过向量加法添加到F的隐藏层。状态预测网络F进而被构建为从电压状态Vi
到容量Qi的简单函数映射,通过对F求解自动微分可以获得dQ/dV信息并添加到网路训练中。
] 作为输入,通过向量加法添加到F的隐藏层。状态预测网络F进而被构建为从电压状态Vi
到容量Qi的简单函数映射,通过对F求解自动微分可以获得dQ/dV信息并添加到网路训练中。【应用实例-1:从实验数据出发重构Mn-DRX设计思路】

我们展示了几个例子来说明DRXNet如何学习潜在的正极化学并协助设计新材料。作为高丰度过渡金属元素,Mn在设计下一代正极材料方面备受关注。Lun等人提出了基于Mn-DRX的三个主要设计自由度:(1) Li含量,控制促进Li扩散的渗流网络;(2) Mn含量,在低Mn量下实现高容量需要大量的氧离子参与氧化还原反应,导致循环性能差;(3) F含量,降低总阳离子价态,进而提供更大的自由度来优化Li和Mn含量。这些设计思路相互之间高度关联,并对性能产生非线性影响。我们使用DRXNet预测DRX化合物在1.5至4.8 V之间、20 mA/g电流速率下第1和第30个循环的放电容量,并将结果作为Li和F含量的函数显示在图B和C中。DRXNet很好地捕捉到了F对性能的影响,这在实验上得到了广泛的验证:更高的F含量导致第一个循环的放电容量降低,但第30个循环的容量升高。这与其在促进表面稳定性方面的作用一致。特别是,阳离子无序的Li2MnO2F被预测在第一个循环具有最高容量( > 320 mAh/g),但在第30个循环具有最低容量,这与文献中所报道的实验观察结果一致。为了展示DRXNet外推能力,图B和C中用蓝星标出了训练数据集中的组分。由此可以观察到,即使在成分图上训练点的分布有限,DRXNet也能提供较为合理一致的预测。由于DRXNet在Li-Mn-O-F化学空间之外的各种成分上同样进行训练,因此可以将其归因于从其他含F和不含F化合物的迁移学习。该例子展示了如何从从数据驱动的角度,结合实验数据挖掘进行总结、推广设计准则。高熵DRX一般由许多元素组成,为电池材料发现提供了广阔的化学空间。我们考虑了来自二价金属Mn2+, Fe2+, Ni2+, Mg2+ 和三价金属Mn3+, Cr3+, V3+, Fe3+,并使用DRXNet进行虚拟高通量筛选。我们展示了两个预测的高熵DRX:
如图所示,我们利用DRXNet预测了两种高熵材料在不同电流密度下的电化学性能。对于HE-1,DRXNet预测在20 mA/g电流速率下的放电容量超过270 mAh/g。该化合物在接近3V时输出最大放电容量,并在3V以下转变为更高的电压斜率,这是基于Mn和Cr氧化还原的特征,在其他DRX中也被广泛观察到。HE-1在放电时具有异常高的倍率性能,在1000 mA/g下的估计容量接近200 mAh/g。之前的工作表明,多元素替代(即高熵策略)会阻碍形成导致Li动力学不良的短程序结构,进而提高倍率性能。此外,Cr的掺入及其在高电压下迁移为Li输运创造了更大的0-TM渗流网络。这两个特征都改善了Li扩散动力学,DRXNet清楚地学习了这些特征,并合理地外推到高熵成分的电化学性能预测中。作为与HE-1的比较,我们用部分V3+替代Cr3+构造了HE-2。DRXNet很好地捕捉到了由于V5+/ V3+还原的低电位导致的电压曲线形状变化。结果清楚地表明,随着V3+的掺入,在低电压区域可以观察到近乎恒定的斜率,这与已报道的基于V的DRX正极的特征相类似。DRXNet预测HE-2在1000 mA/g时保持近170 mAh/g的容量,优于迄今为止报道的大多数DRX正极材料。DRXNet代表了电池材料研究中开发机器学习模型的重要进展,展示了一种从实验数据角度出发的新思路。通过收集过去近五年DRX正极的电化学数据,研究人员建立了一个包含14种不同金属元素的多样化数据集,其中包括超过19,000条不同化学组分和条件下的放电电压曲线。借助这一大规模数据集的学习,DRXNet模型能够准确捕捉DRX正极在各种条件下循环曲线的关键特征,例如在Li-Mn-O-F等化学体系中的广泛适用性,并对几种实验上尚未探索的高熵体系做出了预测(文中所展示的电化学性能均为机器学习模型预测结果)。
作为在多化学组分、不同实验参数上训练的通用模型,DRXNet为快速识别新型正极材料提供了数据驱动的解决方案。通过不断完善模型并结合额外的数据和参数,我们预计这种机器学习框架将在发现和优化下一代电池材料方面发挥越来越关键的作用,为加速清洁能源和碳中和技术发展开辟了新的途径。我们将该模型代码、预训练模型 、和大部分训练数据开源,以促进电化学、科学计算及AI for Science研究领域的协同发展Peichen Zhong, Bowen Deng, Tanjin He, Zhengyan Lun, and Gerbrand Ceder, “Deep learning of experimental electrochemistry for battery cathodes across diverse compositions”, Joule 8, 1–18 (2024) https://doi.org/10.1016/j.joule.2024.03.010











