Mime1为构建基于机器学习的集成模型提供了一个用户友好的解决方案,利用复杂的数据集来识别与预后相关的关键基因。
前面单独介绍了Lasso ,randomForestSRC,Enet(Elastic Net),CoxBoost 和 SuperPC 构建生存模型的方法和参数,本文介绍如何使用Mime1包一体式完成文献中的101种机器学习组合的分析,并输出文献级别的图。
除此以外额外介绍一下(1)替换自己数据时注意的点 (2)如何提取指定模型下的riskscore结果进行后续分析 和 (3)如何对目标癌种进行模型比较(胶质瘤可以使用数据内置的)(个人认为更重要)
该包集合了10种机器学习的包,所以安装上会稍微繁琐一下,给点耐心缺什么下载什么。
# options("repos"= c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))# options(BioC_mirror="http://mirrors.tuna.tsinghua.edu.cn/bioconductor/")if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
depensfor(i in 1:length(depens)){ depen if (!requireNamespace(depen, quietly = TRUE)) BiocManager::install(depen,update = FALSE)}
if (!requireNamespace("CoxBoost", quietly = TRUE)) devtools::install_github("binderh/CoxBoost")
if (!requireNamespace("fastAdaboost", quietly = TRUE)) devtools::install_github("souravc83/fastAdaboost")
if (!requireNamespace("Mime", quietly = TRUE)) devtools::install_github("l-magnificence/Mime")
library(Mime1)
首先查看示例数据
1. 基因集数据
可以是差异基因,热点通路(MsigDB),WGCNA或者PPI找到的hub gene ,或者某种单细胞亚型的marker gene 等等任何方式得到的目标基因集
load("./External data/genelist.Rdata")#> [1] "MYC" "CTNNB1" "JAG2" "NOTCH1" "DLL1" "AXIN2" "PSEN2" "FZD1" "NOTCH4" "LEF1" "AXIN1" "NKD1" "WNT5B" #>[14] "CUL1" "JAG1" "MAML1" "KAT2A" "GNAI1" "WNT6" "PTCH1" "NCOR2" "DKK4" "HDAC2" "DKK1" "TCF7" "WNT1" #>[27] "NUMB" "ADAM17" "DVL2" "PPARD" "NCSTN" "HDAC5" "CCND2" "FRAT1" "CSNK1E" "RBPJ" "FZD8" "TP53" "SKP2" #>[40] "HEY2" "HEY1" "HDAC11"
2. 生存数据与基因表达信息
load("./External data/Example.cohort.Rdata") # 生存数据与基因表达信息list_train_vali_Data[["Dataset1"]][1:5,1:5]# ID OS.time OS MT-CO1 MT-CO3#60 TCGA.DH.A66B.01 1281.65322 0 13.77340 13.67931#234 TCGA.HT.7607.01 96.19915 1 14.96535 14.31857#42 TCGA.DB.A64Q.01 182.37755 0 13.90659 13.65321#126 TCGA.DU.8167.01 471.97707 0 14.90695 14.59776#237 TCGA.HT.7610.01 1709.53901 0 15.22784 14.62756
其中list_train_vali_Data是含有2个数据集的列表,每个数据集的第一列为ID ,2-3列为生存信息(OS.time ,OS) ,后面为基因表达量。
1. 构建101机器学习模型组合
该包大大降低了学习成本,可以通过ML.Dev.Prog.Sig函数直接构建
res list_train_vali_Data = list_train_vali_Data, unicox.filter.for.candi = T, unicox_p_cutoff = 0.05, candidate_genes = genelist, mode = 'all',nodesize =5,seed = 5201314 )
ML.Dev.Prog.Sig() 可选 all, single 和 double三种模式. all 为所有10种算法 以及 组合 . single 为用10种算法中的一种. double 为两种算法的组合,一般情况下使用 all 模式.
默认情况下 unicox.filter.for.candi 为 T , 会先对训练集进行单因素cox分析,unicox_p_cutoff 显著的基因会用于构建预后模型.
如果使用自己数据的时候,需要注意:
(1)替换自己数据注意前三列的要求,且将多个数据集以列表形式存储。
(2)分析之前最好先确认 所有数据集中是否 有基因集列表中的所有基因 ,减少报错。
(3)种子数确定好,会有一些小的影响 。
好奇查看后发现示例数据Dataset2中缺少基因集中的几个基因,但是为什么没有报错呢 ?
data2 % dplyr::select(ID , OS.time , OS, genelist )#Error in `dplyr::select()`:#! Can't select columns that don't exist.#✖ Columns `JAG1`, `DKK4`, and `WNT1` don't exist.#Run `rlang::last_trace()` to see where the error occurred.
通过View(ML.Dev.Prog.Sig) 检查函数,设置unicox.filter.for.candi = T 后会先做单因素cox分析,单因素显著的基因 才会作为机器学习101模型组合的候选基因进行后续分析,而下图红框中单基因显著的基因,恰好没有dataset2中缺少的`JAG1`, `DKK4`, and WNT1 基因,因此没有报错。
但是在不确定哪些基因单因素预后显著的前提下,分析自己数据时候还是先确保训练集和验证集均有基因列表的所有基因。
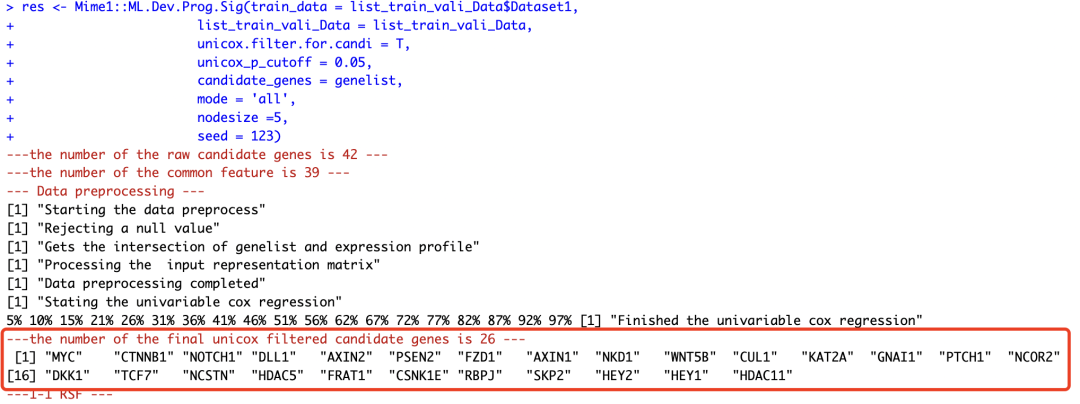
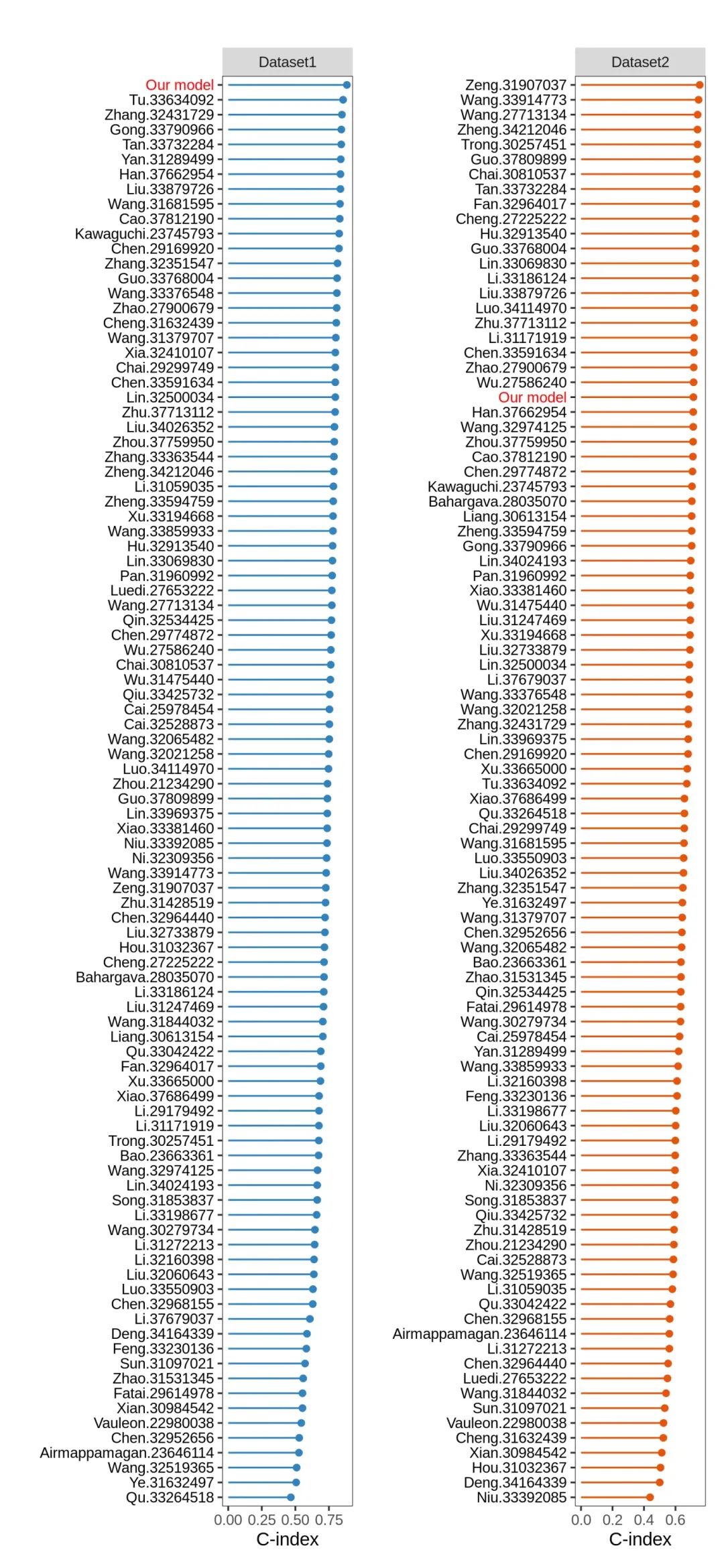
2. C-index 展示
示例数据list_train_vali_Data 为2个数据集的list,结果图中队列为2个,最后两列为Cindex的均值,这也就是机器学习模型组合文献中的主图。
cindex_dis_all(res, validate_set = names(list_train_vali_Data)[-1], order = names(list_train_vali_Data), width = 0.35)
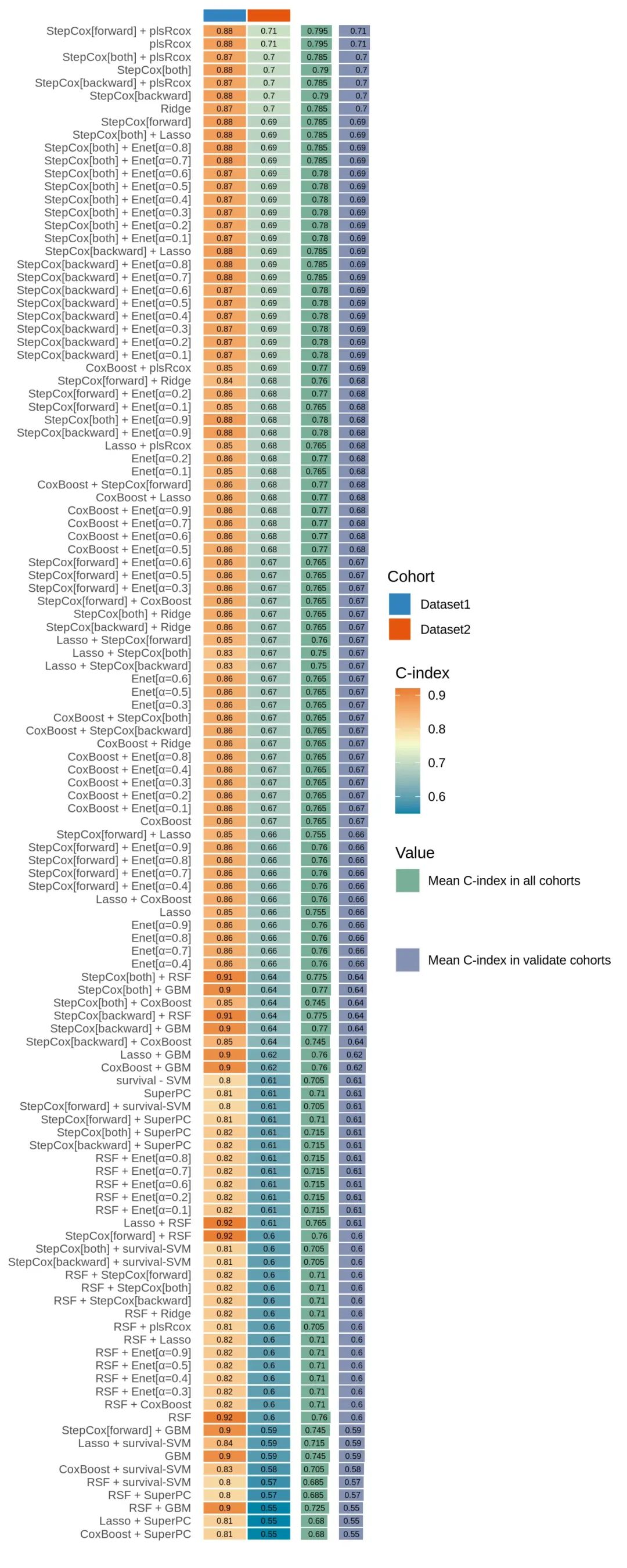
3. 查看指定模型的结果
假设我们选择第一个模型(StepCox[forward] + plsRcox) ,可以单独查看该模型下各个数据集的cindex表现
cindex_dis_select(res, model="StepCox[forward] + plsRcox", order= names(list_train_vali_Data))
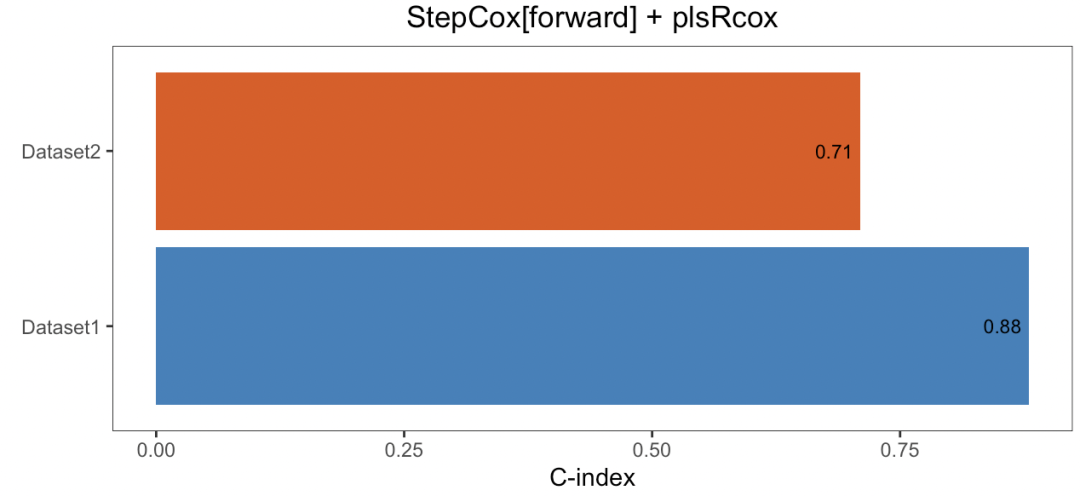
也可以查看该模型下各个数据集的KM曲线情况
survplot for (i in c(1:2)) { print(survplot[[i]] dataset = names(list_train_vali_Data)[i], #color=c("blue","green"), median.line = "hv", cutoff = 0.5, conf.int = T, xlab="Day",pval.coord=c(1000,0.9)) )}aplot::plot_list(gglist=survplot,ncol=2)
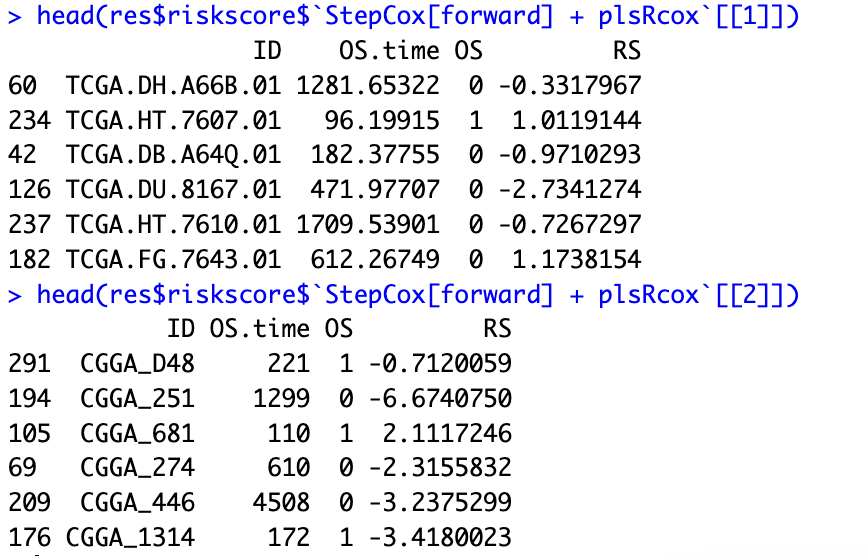
提取模型RS结果
这里有个很重要的点是要提取指定模型下的RS结果,然后就可以根据自己的需求重新绘制KM 以及 独立预后分析,森林图,列线图等其他分析了。
结果都在res中,根据str(res)知道对应的信息,提取即可
head(res$riskscore$`StepCox[forward] + plsRcox`[[1]])head(res$riskscore$`StepCox[forward] + plsRcox`[[2]])
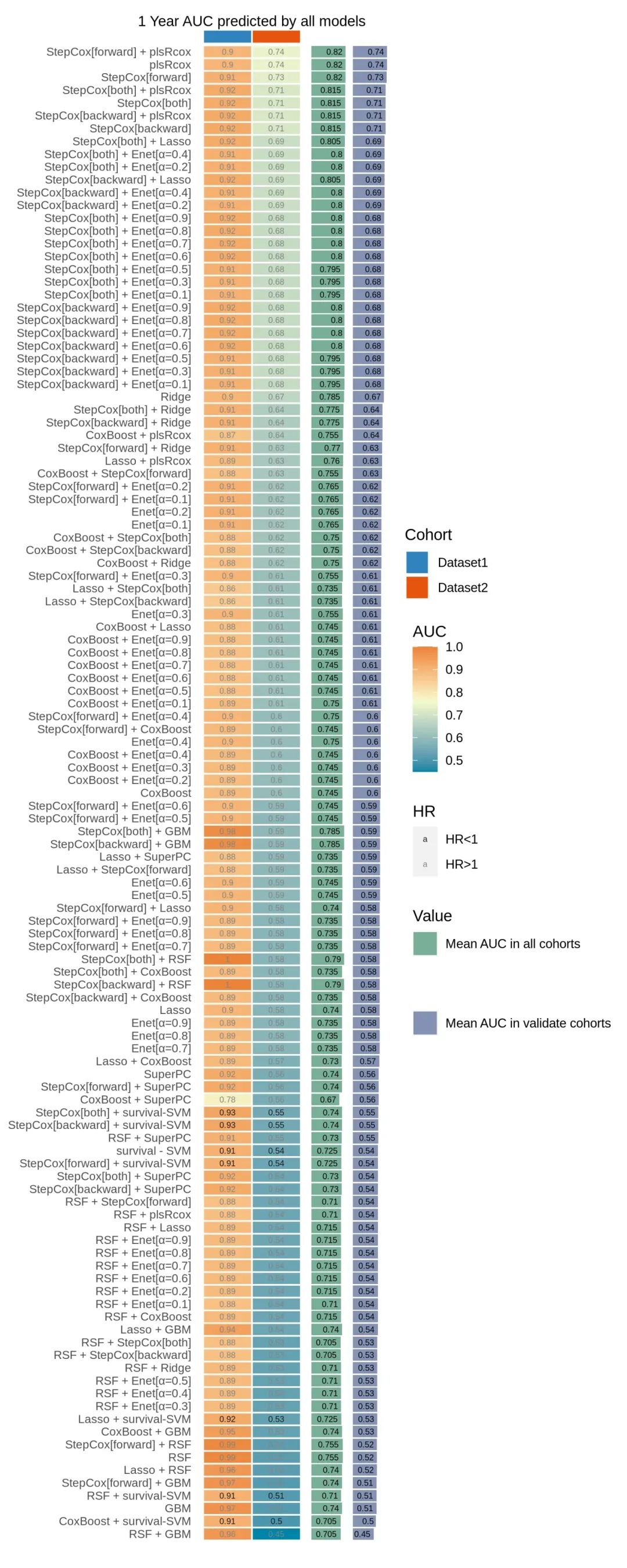
4. AUC结果
计算每个模型的1年,3年,5年 的 auc值 ,并可视化所有模型的1年auc结果
all.auc.1y inputmatrix.list = list_train_vali_Data,mode = 'all',AUC_time = 1, auc_cal_method="KM")all.auc.3y inputmatrix.list = list_train_vali_Data,mode = 'all',AUC_time = 3, auc_cal_method="KM")all.auc.5y inputmatrix.list = list_train_vali_Data,mode = 'all',AUC_time = 5, auc_cal_method="KM")
auc_dis_all(all.auc.1y, dataset = names(list_train_vali_Data), validate_set=names(list_train_vali_Data)[-1], order= names(list_train_vali_Data), width = 0.35, year=1)
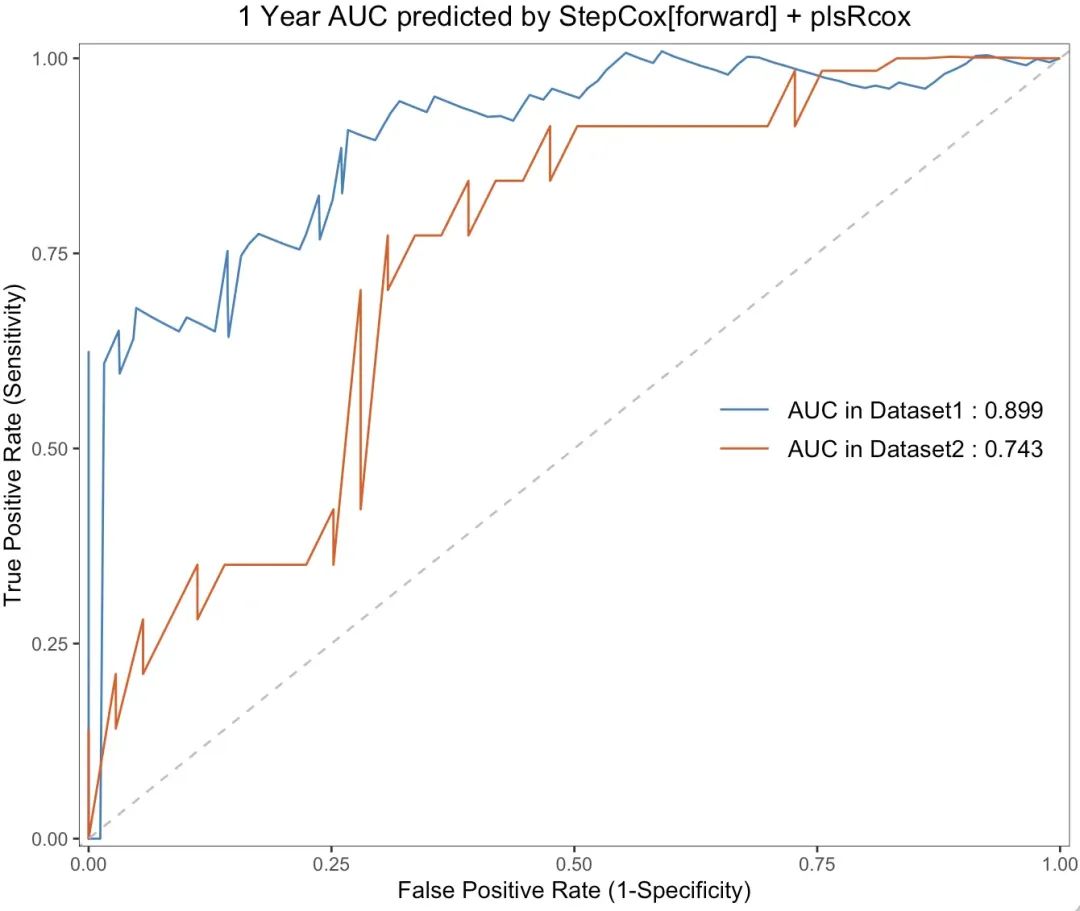
同样可以绘制选定模型下的auc曲线
roc_vis(all.auc.1y, model_name = "StepCox[forward] + plsRcox", dataset = names(list_train_vali_Data), order= names(list_train_vali_Data), anno_position=c(0.65,0.55), year=1)
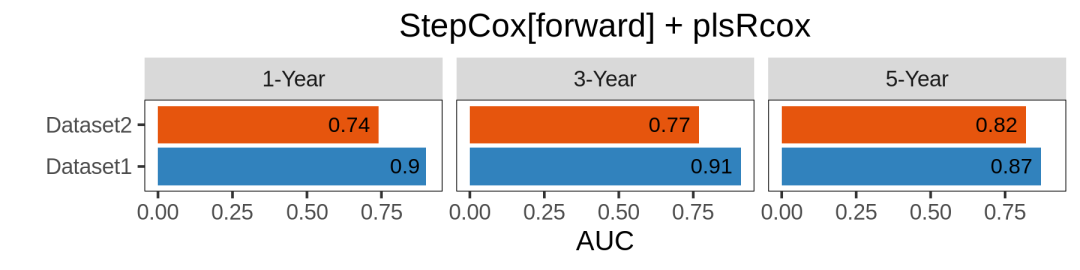
auc_dis_select(list(all.auc.1y,all.auc.3y,all.auc.5y), model_name="StepCox[forward] + plsRcox", dataset = names(list_train_vali_Data), order= names(list_train_vali_Data), year=c(1,3,5))
5. 模型比较
该包还提供了和之前文献报道的预后模型比较的函数,当然只提供了胶质瘤的。
那如果你做的是其他癌种呢?可以通过查看函数了解是怎样的输入形式,然后就做对应的替换后就可以分析了。(很重要)
cc.glioma.lgg.gbm type.sig = c('Glioma','LGG','GBM'), list_input_data = list_train_vali_Data)cindex_comp(cc.glioma.lgg.gbm, res, model_name="StepCox[forward] + plsRcox", dataset=names(list_train_vali_Data))
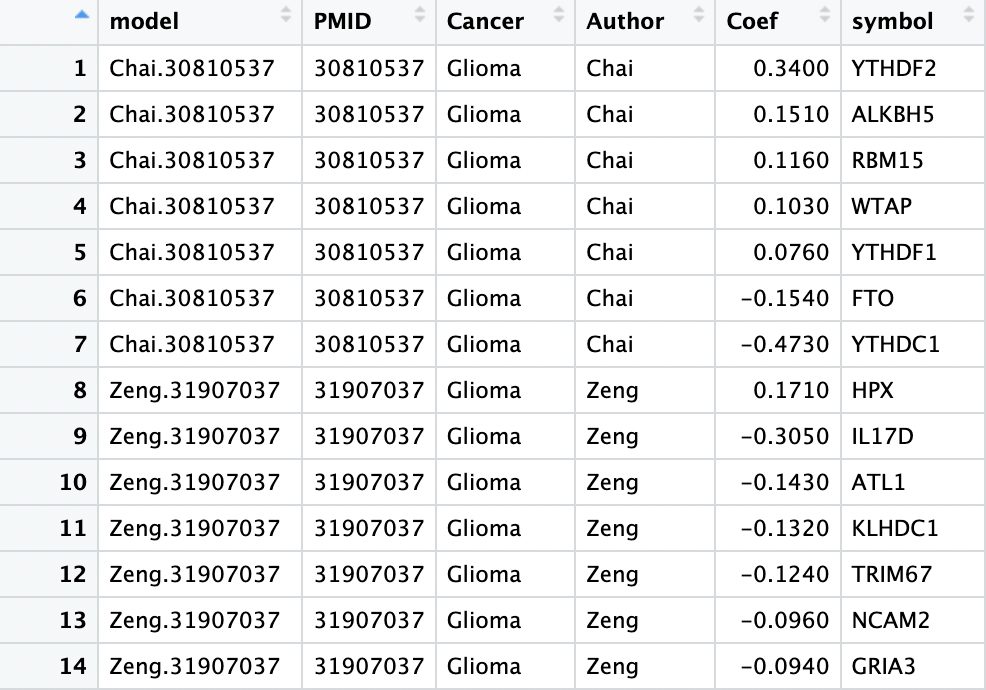
查看函数,找到内置模型的形式
type.sig = c('Glioma','LGG','GBM')pre.prog.sig if (all(type.sig %in% names(pre.prog.sig))) { if (length(type.sig) == 1) { sig.input } else { sig.input for (i in 2:length(type.sig)) { sig.input } } } View(sig.input)
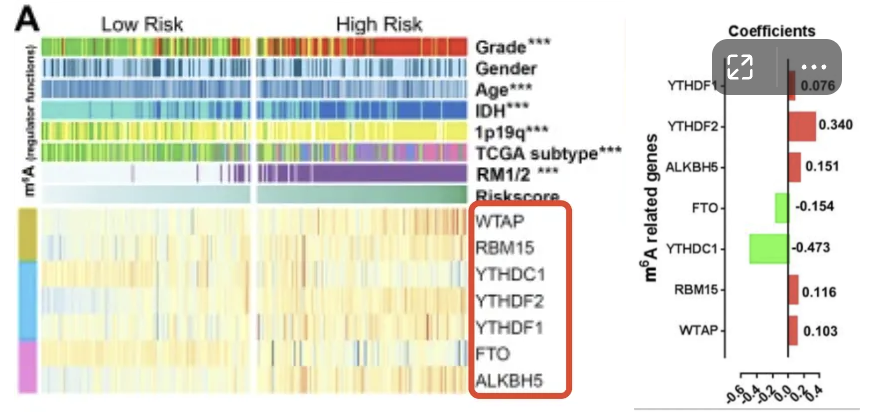
查看30810537文献,发现最终构成riskscore的基因(热图)与 该包内置的基因一致,右图的系数也一致。
OK ,到这里就完成了预后模型的构建以及验证,后面可能还需要根据文中的内容将模型RS提取出来进行独立预后检验以及一些可视化分析。
现在你要做的就是 (1)准备目标癌种的TCGA数据和GEO数据--用于构建预后101模型 ,(2)目标癌种预后模型的基因列表 以及 对应的系数 -- 用于模型比较。(3)写文章发表。
参考资料:https://github.com/l-magnificence/Mime?tab=readme-ov-file
示例数据:直接下载上面github的External data文件夹中的文件,或者后台回复“Mime” 获取。
RNAseq纯生信挖掘思路分享?不,主要是送你代码!(建议收藏)










