作者:梅菜
编辑:李宝珠
转载请联系本公众号获得授权,并标明来源
来自日本东京大学医科学研究所的研究团队提出了一种名为 STAIG (基于图像辅助的图对比学习进行空间转录组学分析)的深度学习框架,能够在无需对齐的情况下整合基因表达、空间数据和组织学图像。
生物组织是由多种类型细胞构成的复杂网络,这些细胞通过特定的空间配置执行重要功能。近年来,10x Visium、Slide-seq、Stereo-seq 和 STARmap 等空间转录组学 (ST) 技术的进步,使得生物学家们能够在空间结构内绘制基因数据,从而各类疾病提供更深入的见解。不过,ST 技术严重依赖于识别具有统一基因表达和组织学特征的空间区域。目前,主要有两种识别方法:非空间聚类和空间聚类方法,非空间聚类方法仅基于基因表达进行聚类,往往导致聚类结果不连贯;空间聚类方法使用图卷积模型整合基因和空间信息,但在将 ST 数据转换为图结构时依赖人为定义的距离标准,可能引入偏差;同时,利用组织学图像的方法也面临挑战,因为其易受染色质量变化的影响;此外,大多数现有方法的批次整合仍需手动干预,例如手动对齐坐标或依赖额外工具。
为了克服这些挑战,来自日本东京大学医科学研究所的研究团队提出了一种名为 STAIG (基于图像辅助的图对比学习进行空间转录组学分析)的深度学习框架,能够在无需对齐的情况下整合基因表达、空间数据和组织学图像。
STAIG 通过自监督模型从苏木精和伊红 (H&E) 染色图像中提取特征,无需依赖大规模组织学数据集进行预训练。此外,STAIG 在训练过程中动态调整图结构,并利用组织学图像信息选择性排除同源负样本,从而减少初始构建带来的偏差。
最后,STAIG 通过局部对比识别基因表达共性,实现端到端的批次整合,无需手动坐标对齐,并有效减少批次效应。研究人员在多个数据集上评估了 STAIG,结果表明其在空间区域识别方面具有良好性能,并能揭示肿瘤微环境中的详细空间和基因信息,促进对复杂生物系统的理解。
相关成果以「STAIG: Spatial transcriptomics analysis via image-aided graph contrastive learning for domain exploration and alignment-free integration」为题,发表于 Nature Communications。
研究亮点:
* STAIG 模型能够在无需预对齐的情况下整合组织切片,并消除批次效应* STAIG 模型适用于从不同平台获取的数据,无论是否包含组织学图像* 研究人员证明了 STAIG 能够高精度识别空间区域,并揭示肿瘤微环境的新见解,展现出其在解析空间生物复杂性方面的广阔潜力
论文地址:
https://www.nature.com/articles/s41467-025-56276-0
关注公众号,后台回复「STAIG」获取完整 PDF该研究所用数据集下载地址:
https://go.hyper.ai/m5YC4
数据集:收集 ST 数据集和来自不同平台的组织学图像
研究人员下载了公开可用的 ST 数据集和来自不同平台的组织学图像,如下图所示。ST 数据集包含人脑背外侧前额叶皮层 (DLPFC) 数据集、人类乳腺癌数据集、鼠脑数据集、Slide-seqV2 数据集、STARmap 数据集等。
数据集下载地址:
https://go.hyper.ai/m5YC4
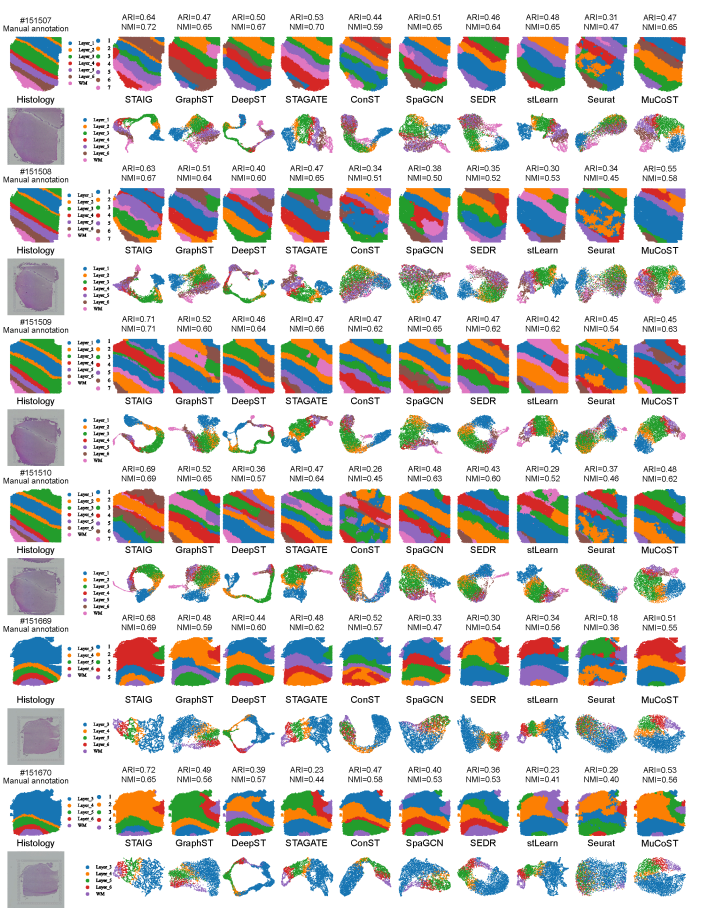
H&E 染色图像、人工注释,以及 STAIG 与基线方法
* 来自 10x Visium 平台的人脑背外侧前额叶皮层 (DLPFC) 数据集包含来自 3 名个体的 12 个切片,每名个体提供 4 个间隔为 10μm 和 300μm 的切片,每个切片的点计数在 3,498 到 4,789 之间,这些切片已被手动标注为皮层层 L1–L6 及白质 (WM);
* 鼠脑数据集包括前后两个切片,分别包含 2,695 和 3,355 个点;
* 对于斑马鱼黑色素瘤,研究人员分析了切片 A 和 B,分别包含 2,179 和 2,677 个点;
* 对于集成实验,使用了 DLPFC 和鼠脑数据集,鼠嗅球的 Stereo-seq 数据集包含 19,109 个点,分辨率为 14μm;
* Slide-seqV2 数据集具有 10μm 分辨率,包括鼠海马 (来自中央四分之一半径的 18,765 个点) 和鼠嗅球 (19,285 个点);
* STARmap 数据集包含 1,207 个点;
* 对于 MERFISH 数据集,人类 MTG 包含 3,970 个点,而鼠 1 和鼠 2 的 VIS 区域分别包含 5,995 和 2,479 个点。模型架构:基于图像辅助的图对比学习进行空间转录组学分析
下图描述了 STAIG 的整体框架,这是一种使用图对比学习结合高性能特征提取来整合基因表达、空间坐标和组织学图像的深度学习框架,包含 6 个模块:
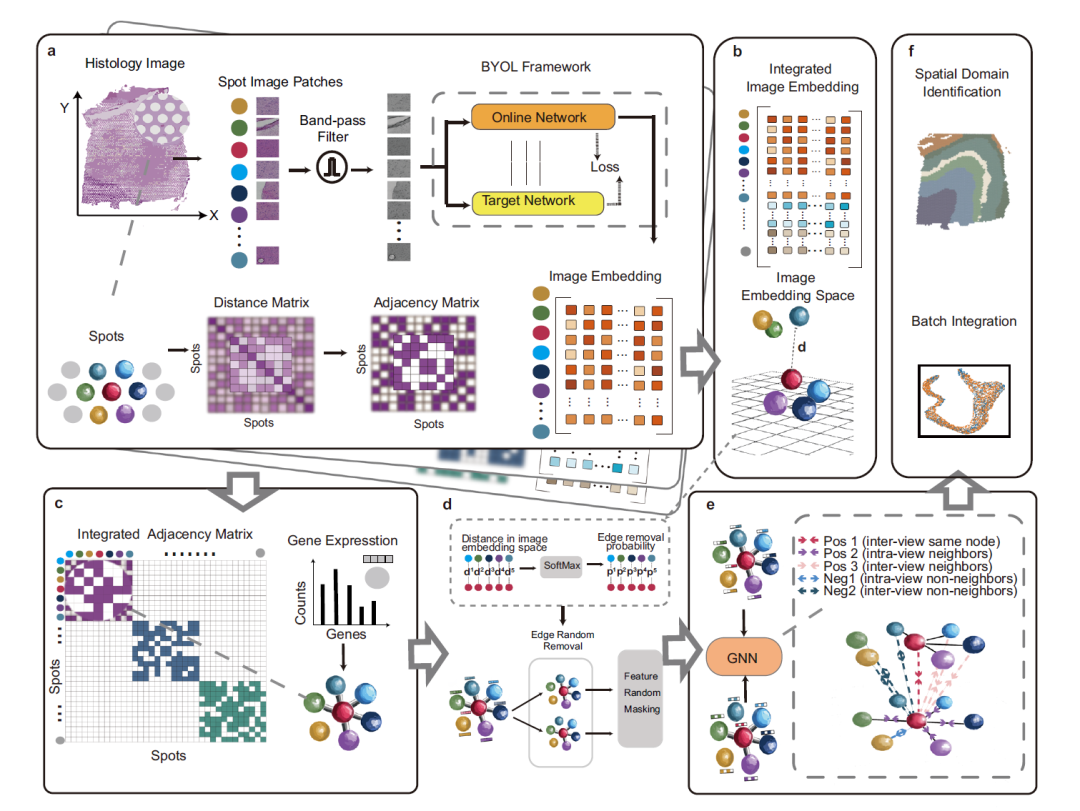
STAIG 框架概述
首先,如下图 a 部分,为了减少噪声和组织染色不均的影响,STAIG 首先将组织学图像分割成与数据点空间位置对齐的小块 (Spot Image Patches),随后使用带通滤波器 (Band-pass Filter) 对图像进行优化处理,图像嵌入特征通过 Bootstrap Your Own Latent (BYOL) 自监督模型提取,同时基于数据点之间的空间距离构建邻接矩阵。
每个切片都包括带有空间坐标的斑点、基因数据和可选的苏木精和伊红染色图像
如下图 b 部分,为了整合不同组织切片的数据,STAIG 采用纵向堆叠(stacking) 的方式,将多个组织切片的图像嵌入特征整合。
如下图 c 部分,各切片的邻接矩阵采用对角放置 (diagonal placement) 方法合并,形成整合后的邻接矩阵,随后,该矩阵用于构建图结构,其中基因表达数据作为节点信息。
每个部分的邻接矩阵按对角线方向组合,形成一个综合邻接矩阵
如下图 d 部分,对于通过边相连的测点,在图像嵌入空间中计算其距离,并利用 SoftMax 函数将这些距离转换为随机边移除的概率,原始图在此基础上经历两轮随机边移除 (Edge Random Removal),从而生成两个增强视图,接着,对这些视图中的节点特征进行随机掩码处理。
然后,如图 e 部分,增强视图随后通过共享的图神经网络 (GNN) 进行处理,并受邻域对比目标 (neighboring contrastive objective) 的引导,该方法旨在在两个图视图中拉近相邻节点之间的距离,同时拉远非相邻节点。
最终,如图 f,训练后的 GNN 生成嵌入,以识别空间区域并最小化连续组织切片间的批次效应。
利用 GNN 得出的嵌入结果进行空间域识别和整合研究结果:STAIG 在各种条件下均表现出优越性能
研究团队进行了广泛的基准评估,将 STAIG 与其他最先进的 ST 技术进行了比较。结果显示,STAIG 在各种条件下均表现出优越的性能。
脑区识别性能评估
为了评估 STAIG 在组织区域识别中的性能,研究人员将 STAIG 与现有方法进行了对比,包括 Seurat、GraphST、DeepST、STAGATE、SpaGCN、SEDR、ConST、MuCoST 和 stLearn,性能评估指标包括:
* 调整兰德指数 (ARI) 和归一化互信息 (NMI)(用于手动标注的数据集)。
* 轮廓系数 (SC) 和戴维斯-鲍丁指数 (DB)(用于其他数据集)。
① 人脑数据集的表现
整体而言,STAIG 在人脑数据集中表现最佳,取得最高的中位数 ARI (0.69) 和 NMI (0.71),如下图所示:
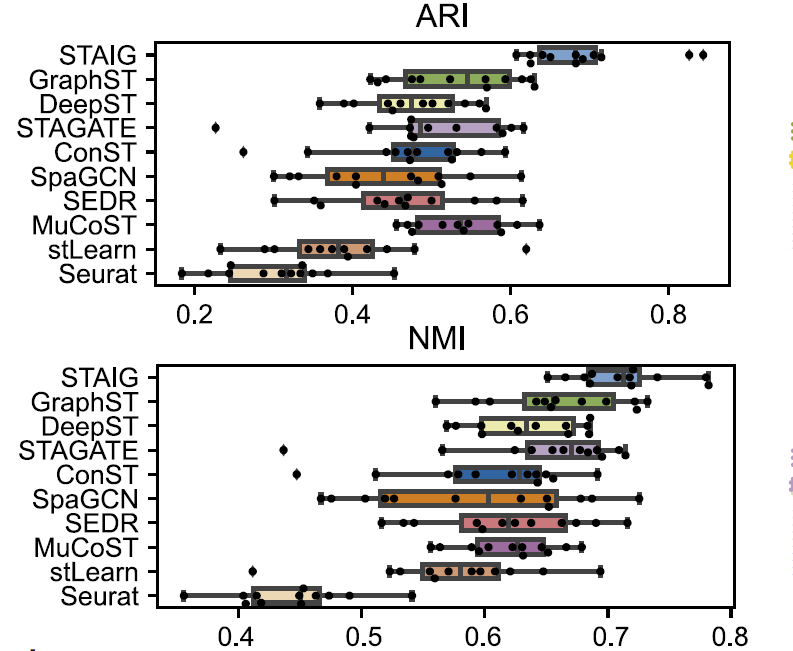
9 种方法在 12 个 DLPFC 切片上的调整兰德指数 (ARI) 和归一化互信息 (NMI) 箱线图
相比之下,现有方法的表现较差:stLearn 误判了一些测点,并遗漏了部分层次;GraphST 的 ARI 为 0.64,NMI 为 0.73,但在 L4 和 L5 层的位置存在较大偏差;其他方法的 ARI 介于 0.25–0.57,NMI 介于 0.42–0.69,主要由于层次比例识别不准确。
② 小鼠脑数据集的表现
如下图,在小鼠后脑数据集中,STAIG 成功识别小脑皮层和海马区,并进一步区分了 Ammon 角 (CA) 和齿状回 (dentate gyrus),与 Allen 小鼠脑图谱注释高度一致;尽管缺少手动标注,STAIG 仍取得最高 SC (0.31) 和最低 DB (1.11),表明其优越的聚类性能。
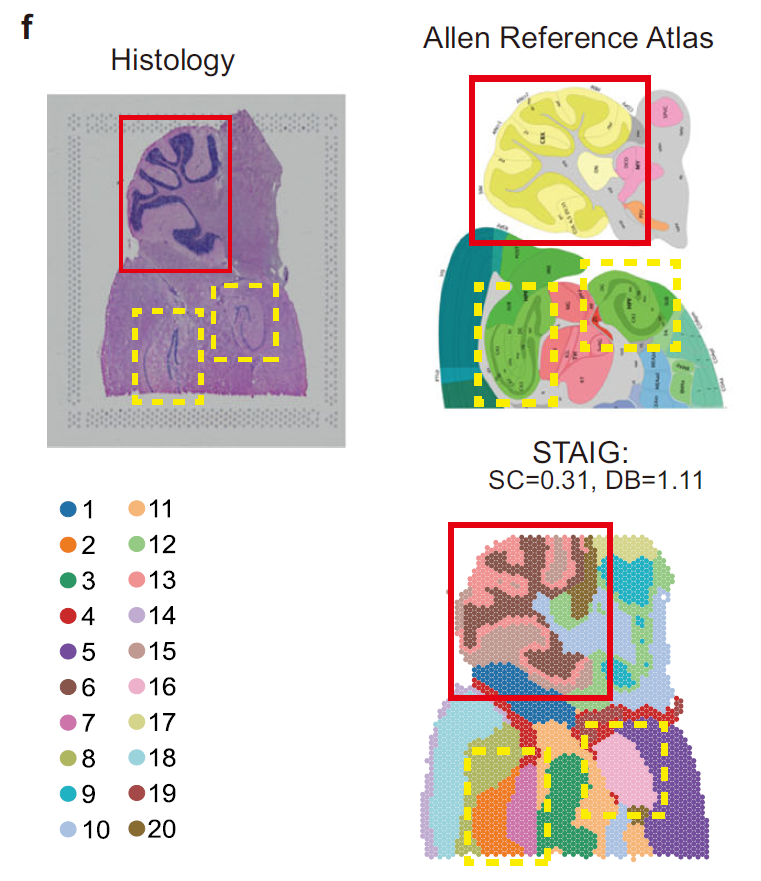
小鼠脑后部组织的 H&E 染色图像、Allen 参考图谱的解剖学标注,以及 STAIG 的聚类结果
又如下图,在小鼠前脑数据集中,STAIG 精确分割了嗅球 (olfactory bulb) 和背侧苍白球 (dorsal pallium),在参考 Long 等人的手动标注后,其 ARI 达 0.44,NMI 达 0.72,均为最高值。
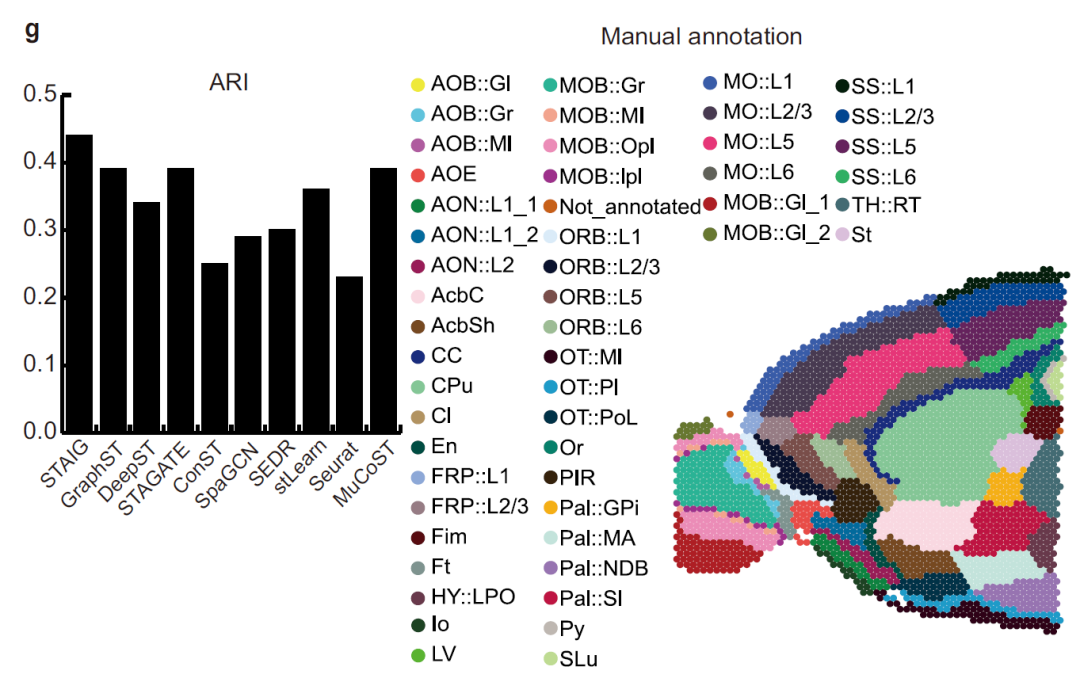
Long 等人的手动标注以及 STAIG 和基线方法在小鼠前部组织上的 ARI 柱状图
图像特征提取的有效性
为了探讨图像特征的影响,研究人员使用 KNN 算法对比了 STAIG 与其他方法 (stLearn、DeepST 和 ConST) 提取的图像特征。
① 大脑组织切片分析
以切片 #151507 为例,如下图,stLearn 的图像特征受染色强度影响严重,导致与真实层次标注不匹配;DeepST 和 ConST 虽采用深度学习,但未能准确捕捉脑组织的复杂纹理特征;STAIG 的特征提取结果与手动标注的层次高度匹配,尽管部分边界仍略显模糊,但几乎不受染色差异的影响。
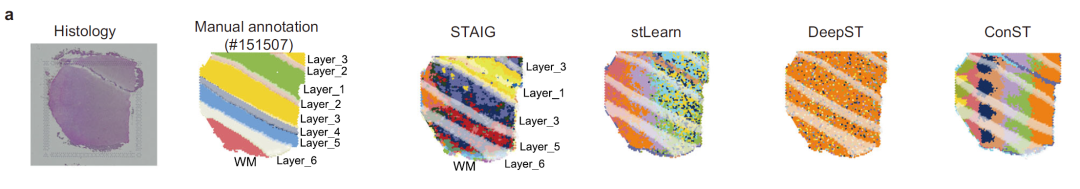
LPFC 切片 #151507 的 H&E 染色图像、手动注释 及 基于图像特征的 KNN 聚类结果,对比 STAIG 与 3 种基于图像的方法 (stLearn、DeepST、ConST)
② 乳腺癌组织图像分析
研究人员进一步使用人类乳腺癌 H&E 染色图像测试图像特征提取能力,如下图所示。
结果显示:stLearn 的图像特征混合了肿瘤和正常区域,区分度较差;ConST 看似将图像分割成不同区域,但放大观察后,区域边界与手动标注存在较大偏差;DeepST 未能提取有效的图像特征;STAIG 精准识别肿瘤区域,其空间聚类结果保持了高度的区域连贯性,且分割区域几乎完美匹配手动标注的轮廓,验证了其卓越的图像特征提取能力。

人类乳腺癌数据集的 H&E 染色图像、基于视觉解读的手动注释及基于图像特征的 KNN 聚类结果,对比 STAIG 与三种基于图像的方法
明确人类乳腺癌 ST 中的肿瘤微环境
在对人类乳腺癌数据集的分析中,研究人员发现 STAIG 的结果与手动注释高度一致,并且达到了最高的 ARI (0.64) 和 NMI (0.70)。值得注意的是,STAIG 提出了稍微不同但更精细的空间分层,特别是对手动注释中的 Healthy_1 区域(下图 a),STAIG 将其细分为子群 3 和 4(下图 b)。
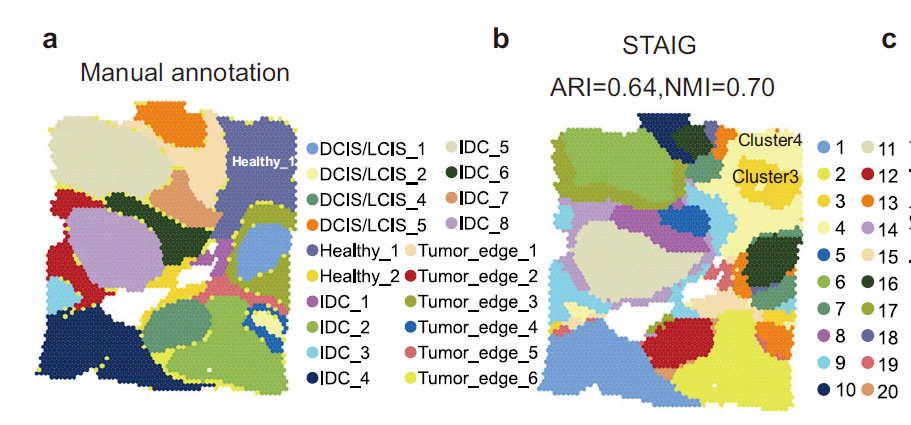
STAIG 的高级空间分析揭示了人类乳腺癌 ST 数据中的癌症相关成纤维细胞 (CAF) 富集群体
总之,通过 STAIG 的多模态整合,研究人员发现子群 3 形成了一个 CAF 密集的肿瘤微环境,并揭示了 CAF 丰富区域的分子特性。
深度学习为 ST 技术发展提供强大工具
随着基因组学和 ST 技术的飞速发展,生物医学界得以探索组织内基因表达的空间分布,从而揭示生物体复杂的功能和结构。ST 技术不仅提供了基因表达的定量信息,还保留了细胞在组织中的空间关系,使相关人员能够研究组织微环境、细胞相互作用以及疾病发生发展的空间特征。然而,由于 ST 数据通常具有高维度、强噪声和批次效应等问题,如何有效地整合和解析这些数据成为当前研究的核心挑战。
深度学习技术,特别是图神经网络 (GNN) 和对比学习方法的引入,为 ST 数据的分析提供了强大的工具。传统的分析方法往往依赖于降维和聚类,而深度学习方法能够自动提取多层次的特征,并通过端到端训练优化数据表示。如前文所述,基于 GNN 的方法可以利用空间邻接信息构建图结构,使模型在捕捉基因表达的同时,也能学习到细胞间的空间依赖关系,而对比学习的引入进一步增强了模型的泛化能力,使其能够在没有标注的情况下学习到关键的空间特征。
除此之外,业界在深度学习+ST 技术结合方面也有不少进展:
2024 年 11 月,中国国家生物信息中心杨运桂团队、中国科学院数学与系统科学研究院张世华团队合作,开发了一种基于深度学习的空间转录组细胞注释工具 STASCAN,通过整合基因表达谱和组织学图像的细胞特征学习来预测组织切片未知区域的细胞类型,并对捕获区域内的细胞进行细分注释,从而大大提高空间细胞分辨率。此外,STASCAN 适用于来自不同 ST 技术的不同数据集,并在破译高分辨率细胞分布和解决增强的组织结构方面显示出显著优势。
该成果以「STASCAN deciphers fine-resolution cell distribution maps in spatial transcriptomics by deep learning」为题,发表在 Genome Biology 上。
* 论文地址:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03421-5
2025 年 1 月 23 日,来自美国普林斯顿大学的研究团队开发了一种全新的深度学习算法 GASTON (Gradient Analysis of Spatial Transcriptomics Organization with Neural networks)。GASTON 通过结合无监督的深度神经网络与可解释性算法,创新性地提出了「等深度 (Isodepth)」的概念,这一概念类似于地形图中的海拔高度,用于量化组织切片中的基因表达空间拓扑结构。、
通过等深度及其梯度,研究人员不仅能够分割组织的不同空间区域,还能识别组织内基因表达的连续变化趋势和关键标志基因。该研究展示了 GASTON 在多种生物样本中的成功应用,包括小鼠大脑、小鼠嗅球、结直肠癌肿瘤微环境等。结果表明,GASTON 可以准确解析组织结构,揭示细胞类型的空间分布与变化规律,同时挖掘出许多在其他方法中被忽视的空间基因表达模式。
相关成果以「Mapping the topography of spatial gene expression with interpretable deep learning」为题发布于 Nature Methods。

* 论文地址:
https://www.nature.com/articles/s41592-024-02503-3
显然,深度学习与 ST 技术的结合不仅提升了数据整合与降噪的能力,还促进了空间生物信息的深入挖掘。未来,随着计算资源的增长和算法的优化,深度学习将在 ST 数据分析中发挥更加重要的作用,为精准医学和个性化治疗提供更有力的支持。
参考资料:
1.https://static-content.springer.com/esm/art%3A10.1038%2Fs41467-025-56276-0/MediaObjects/41467_2025_56276_MOESM1_ESM.pdf
2.https://www.bjqykxy.com/kexueyanjiu/dongwuzhiwu/7361.html
3.https://news.qq.com/rain/a/20250128A057OQ00?suid=&media_id=
4.https://www.medsci.cn/article/show_article.do?id=76f8849301b1










