海归学者发起的公益学术平台
分享信息,整合资源
交流学术,偶尔风月

若想设计一款新的材料比如超强航天合金或高效电池材料,其本质在于破译构成材料的原子在皮秒(万亿分之一秒)与埃米(百亿分之一米)时空尺度下的相互作用密码,这些微观粒子的动态行为直接决定材料的宏观特性。计算机技术的革命性进步,使我们能够构建材料的“数字孪生”系统,通过求解物理方程,推演原子群的运动轨迹与能量状态,预演其性能。这可以有效降低实验研究的时间和经济成本,为高性能材料开发建立高效通道。
势函数,堪称这个数字孪生宇宙的“基础物理法则”,它定义了原子在不同距离、角度下相互作用的能量与受力,从而决定了原子运动的轨迹。开发准确、高效的势函数是原子模拟技术的关键。该领域发展已逾百年,从Lennard-Jones提出经典对势模型起始,至近十余年迎来革命性突破:机器学习方法已被系统性整合于势函数构建。当下,前沿的机器学习势函数模型建立在球谐函数及其张量积构成的复杂数学体系之上。
电子科技大学文明健教授团队创新性地提出“笛卡尔原子矩势函数”(CAMP)。该方法实现完全基于笛卡尔空间中原子的x, y, z坐标构建机器学习势函数,建立更简洁、直观的高效建模体系。其技术核心在于直接通过相邻原子空间坐标构建原子矩(类似于力矩),继而融合多原子位置信息精确捕捉复杂相互作用,并嵌入图神经网络框架形成可系统性迭代优化的势函数(图1)。
图1. CAMP势函数模型
他们在锂电材料、有机分子、水体系及二维石墨烯等多元系统中验证CAMP的卓越性能。在室温下对水进行分子动力学模拟,可以精准预测X射线散射(和中子散射)实验得到的氧-氧径向分布函数(图2)。
图2. CAMP模拟得到水的径向分布函数(RDF)与实验结果对比
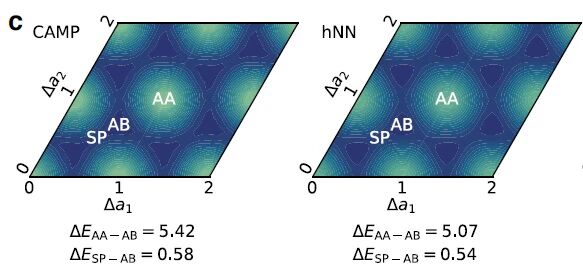
在多层石墨烯材料模拟中,CAMP可以完美复现DFT层间距离改变的能量曲线和层间相对滑移的能量面(图3)。
图3. CAMP在多层石墨烯性能预测中的表现
CAMP计算精度与效率达到或超越主流模型,纳秒级分子动力学模拟保持稳定。该模型为研究人员提供全新工具,其简洁高效的原子相互作用建模范式,有望推动储能材料、电子器件及纳米技术领域的新材料研发进程。该文近期发表于
npj Computational Materials 11:128(2025),英文标题与摘要如下,点击左下角“阅读原文”可以自由获取论文PDF。
Cartesian atomic moment machine learning interatomic potentials
Mingjian Wen*,Wei-Fan Huang, Jin Dai & Santosh Adhikari
Machine learning interatomic potentials (MLIPs) have substantially advanced atomistic simulations in materials science and chemistry by balancing accuracy and computational efficiency. While leading MLIPs rely on representing atomic environments using spherical tensors, Cartesian representations offer potential advantages in simplicity and efficiency. Here, we introduce the Cartesian Atomic Moment Potential (CAMP), an approach to building MLIPs entirely in Cartesian space. CAMP constructs atomic moment tensors from neighboring atoms and employs tensor products to incorporate higher body-order interactions, providing a complete description of local atomic environments. Integrated into a graph neural network (GNN) framework, CAMP enables physically motivated, systematically improvable potentials. The model demonstrates excellent performance across diverse systems, including periodic structures, small organic molecules, and two-dimensional materials, achieving accuracy, efficiency, and stability in molecular dynamics simulations that rival or surpass current leading models. CAMP provides a powerful tool for atomistic simulations to accelerate materials understanding and discovery.