从我们的新课《掌握Python,解锁单细胞数据的无限可能》学习了python版本的功能富集分析,现在就来实践一下~
我们前面分享了GSEApy模块的ORA的功能富集分析方法:python版本的功能富集分析:GSEApy
今天来看看GSEApy模块的GSEA富集分析!
还是老规矩,先给出官网:
GSEApy包官网:https://gseapy.readthedocs.io/en/latest/
差异基因列表
还是使用前面做过的一个芯片数据的差异结果:2万个基因少一半也不影响最后的差异分析富集结果啊?
数据为:https://ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17351,拿到之后进行芯片预处理并做差异表达分析得到一个差异结果,或者 微信找我发你:Biotree123。
或者百度云盘:链接: https://pan.baidu.com/s/1sAXzlxs4jU24ZcAW4LThqQ?pwd=uavp 提取码: uavp 。
读取差异基因结果:
# 导入模块
import pandas as pd
import gseapy as gp
# 读取差异分析结果
# 记住文件路径改成自己的
# index_col=0:第一列读取为行名
deg = pd.read_csv("./GSE17351/DEG.csv", index_col=0)
deg.head()
差异结果如下:
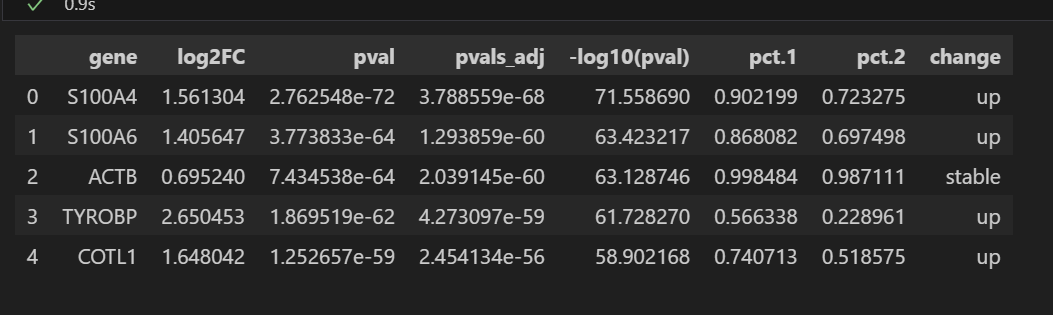
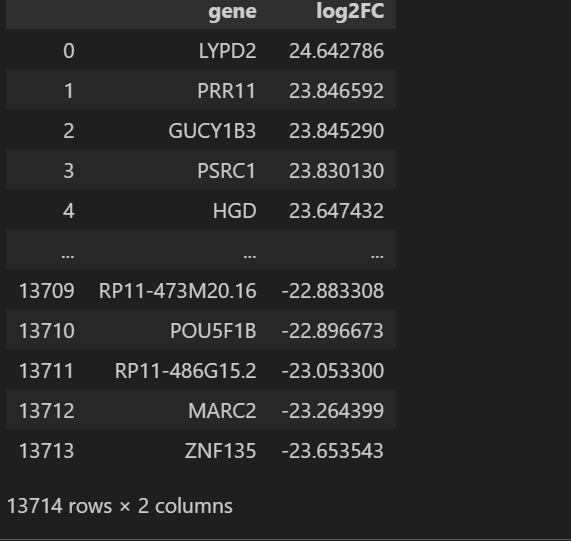
提取基因排序列表
这里用到的基因为全部的基因,包括显著的和不显著的:
ranking = deg[['gene', 'log2FC']]
# 对基因列表排序,降序排列
ranking = ranking.sort_values('log2FC', ascending = False).reset_index(drop = True)
ranking
获取基因集
还是老办法,像之前的稿子一样:python版本的功能富集分析:GSEApy
下载50条 hallmarks 通路:
msig = gp.Msigdb()
gmt = msig.get_gmt(category='h.all', dbver="2024.1.Hs")
# 查看一下基因集合
list(gmt.keys())[0:10]
# 列出某个基因集里的基因
gene_set=gmt['HALLMARK_ADIPOGENESIS']
print(gene_set)

运行GSEA分析
prerank函数:
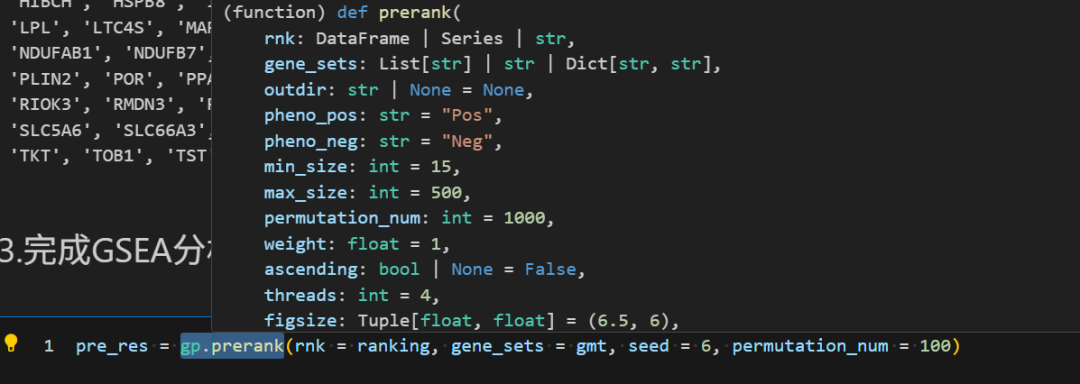
具体参数:
- ranking:输入的排序基因列表,是一个数据框或者Series结构
- gene_sets:输入基因集,是一个list列表或者dict字典结构
- min_size/max_size:基因集大小过滤参数,设置可以让多的基因集留下来
# run gsea
pre_res = gp.prerank(rnk = ranking, gene_sets = gmt, seed = 6, permutation_num = 1000, min_size=1, max_size=10000,outdir="./")
# 结果类型
type(pre_res)
# gseapy.gsea.Prerank
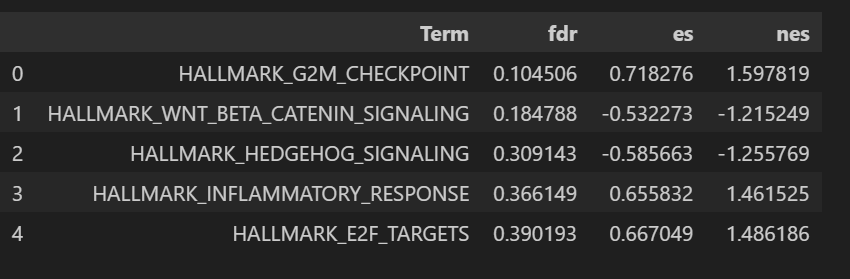
对结果转换为数据框:
out = []
for term in list(pre_res.results):
out.append([term,
pre_res.results[term]['fdr'],
pre_res.results[term]['es'],
pre_res.results[term]['nes']])
out_df = pd.DataFrame(out, columns = ['Term','fdr', 'es', 'nes']).sort_values('fdr').reset_index(drop = True)
out_df.head()
结果可视化
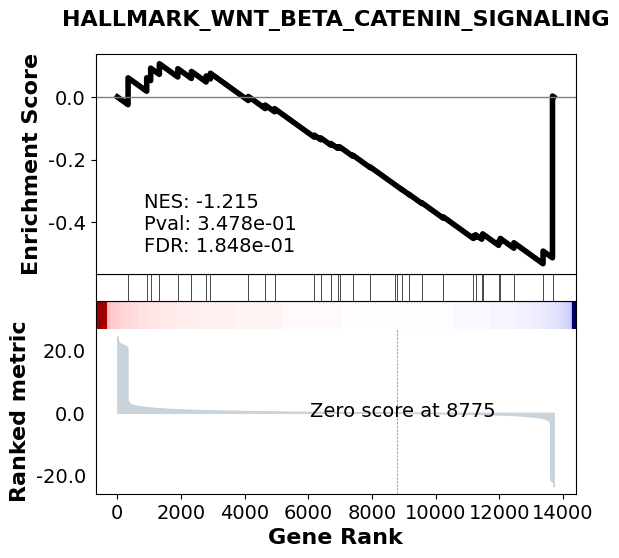
绘制一下GSEA结果经典图:
# 选一条通路看看
# color="black":修改曲线的颜色
gp.gseaplot(rank_metric = pre_res.ranking,term = 'HALLMARK_WNT_BETA_CATENIN_SIGNALING', **pre_res.results['HALLMARK_WNT_BETA_CATENIN_SIGNALING'],color="black")
本次分享到这~
文末友情宣传







